DFT simulation of anatase TiO₂
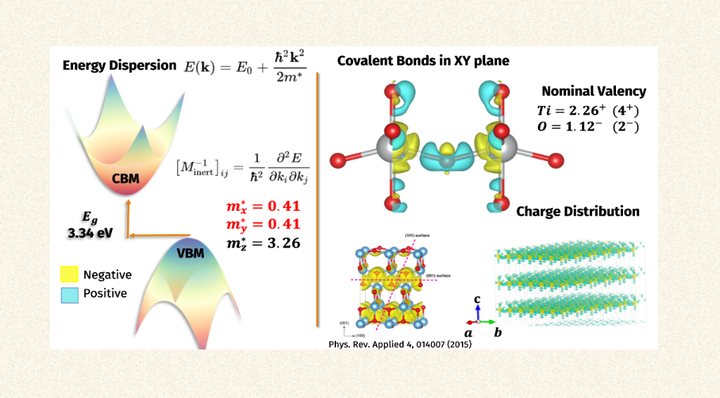 Effective mass of electrons in TiO₂ and their spatial localization.
Effective mass of electrons in TiO₂ and their spatial localization.
Overview
The Superior optical and electronic properties of Titanium Dioxide makes it a promising candidate for designing multifunctional devices tailor-made to use for various applications. Optical properties, especially nonlinear optical properties, of Titanium dioxide changes significantly while it is doped with metals like Silver, Gold and Manganese. Rather than exhibiting a monotonous enhancement or diminution of nonlinear optical properties with respect to an increase in the concentration of the dopant it attains maximum at certain optimal concentrations. First principle simulation of optical properties of Titanium Dioxide, both pristine as well as doped with impurities at different concentrations will help us to predict the composition of the material with superior tailor-made properties before it is tested by experiments.
We carry out Ab-initio simulation of nonlinear optical properties of Titanium Dioxide based on Density Functional Theory(DFT). It is proposed to use different Hierarchy of approximations namely in DFT, Generalized Gradient Approximation(GGA), Hybrid DFT(HSE), to optimize the calculation parameters so that the results can be bench marked with experiments. In addition, in order to overcome the limitations of ground state DFT to calculate properties involving excited states (optical excitation spectra for instance) it requires the use of other computationally intensive advanced techniques that use Bethe-Salpeter Equation (BSE) or GW method. The proposed scheme of complete simulation involves
- Structural optimization(Volume Relaxation) of the system by energy minimization to optimize the structural parameters
- Self-Consistent Field Calculation of relaxed structure
- Calculation of Band structure and optical properties using HSE (Hybrid) Functional to account for the band gap problem inherent in standard GGA-DFT methods
- Quasi-particle Correction to band structure and optical properties of bulk anatase titanium dioxide by performing Hybrid DFT and Green’s function based GW calculations
- Repetition of the above for titanium dioxide system in which metal dopants are incorporated.
We plan to use 5 metal atoms as dopant each of which will be incorporated in the system in five different concentrations making it a set of 25 different computational systems. The average time required per computational system for carrying out the work as mentioned in the proposal is given below showing the detailed break-up over individual jobs. These have been estimated based on the benchmark calculation performed using Vienna Ab-initio simulation Package(VASP) for a super cell containing 50 to 60 atoms.
Sruthil Lal SB
Researcher in Nonlinear Optics
PhD(Physics) in the making| Ultrafast processes and Nonlinear Optics | Jack of some trades, master of none | Master Procrastinator